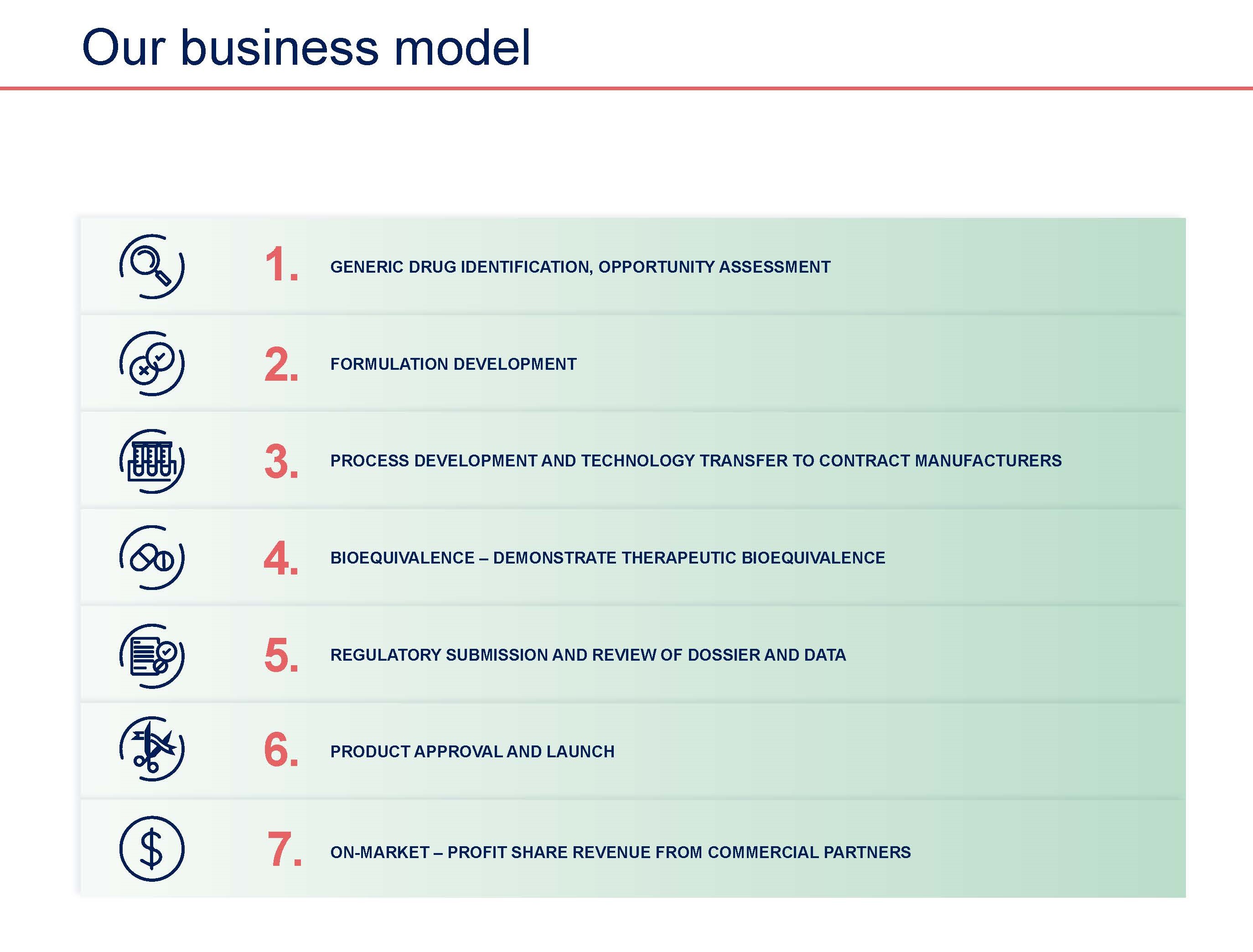
Acrux Generic Product Development
Acrux focuses on developing and commercialising a range of topically applied products with a strong emphasis on complex generic products.
Topical products are applied to the skin as semi-solids (e.g. ointments, creams, gels, lotions, and suspensions) and also include products applied to the ear (otic), nose (nasal), eyes (ophthalmic) and rectum. Acrux uses its internal development capabilities and know-how to develop generics which target a substantial portion of the US topical market. The development time required for generic products is substantially shorter and less costly than the length of time required for a new drug development.
More information on Generic Products
Generic pharmaceutical products are the pharmaceutical and therapeutic equivalents of the brand product. Accordingly, generic products provide a safe, effective and cost-efficient alternative to users of these reference brand.
Generic product development is generally less time-consuming and complex than the new chemical entity development process. It usually does not require new preclinical and clinical studies, because it relies on the studies establishing safety and efficacy conducted for the referenced innovator product, often called the Reference Listed Drug or RLD. It may require one or more bioequivalence studies to show that the generic drug is bioequivalent to the previously approved reference listed brand drug (RLD). On occasions a bioequivalence study waiver (“biowaiver”) may be granted. The RLD is a product that has been previously approved through the respective regulatory agency such as the Food and Drug Administration (FDA) in the United States, the Therapeutic Goods Administration (TGA) in Australia or the European Medicines Agency (EMA). Acrux’s major focus is the FDA approval of its generic product. The FDA application to market a generic drug is called an Abbreviated New Drug Application (ANDA) and once approved, the product is listed in the FDA’s publication popularly known as the Orange Book.
For further information:
https://www.fda.gov/media/107601/download
https://accessiblemeds.org/generic-medicines
www.tga.gov.au/community-qa/generic-prescription-medicines-fact-sheet